Learning Objectives:
- To recognize various different kinds of chemical identifiers used on computers
- How computer systems interpret various kinds of chemical names, formulas, and other identifiers in chemically meaningful ways.
- What kind of information about chemical structure a computer program can and cannot derive from different representations and identifiers.
- How a connection table represents chemical structure
- The factors that you should consider in selecting an appropriate kind of chemical name, formula, or identifier to use, including the information you want to communicate, the kind of chemical entity you’re referring to, the audience with whom you’re communicating, and the medium in which you’re communicating.
Communicating about chemical structure on computers
2.1. Cheminformatics and structure representation
2.2. Identifying chemical compounds on computers
2.3. Translating and exchanging identifiers
2.4. Assignment
2. Communicating about chemical structure on computers
Overview
In Part 1 of this module, you reviewed how chemical names and formulas work, why they work like they do, and how chemists interpret (and misinterpret) them.
In Part 2, we will cover how chemical structure is represented on computers. We will begin by discussing why chemical structure representation is of such central importance to cheminformatics. We will then introduce several chemical identifiers and representations developed specifically for use on computers. In later modules, you’ll learn about these in more detail. But whether or not you’re doing cheminformatics, most chemical communication involves computers in one way or another. We will discuss how that may matter.
Miscommunication is especially apt to arise when you’re translating one kind of representation or identifier for a compound into another. Therefore, we will conclude Part 2 by discussing how to translate between one kind of name or formula and another (and what can get lost in translation).
A reminder: later modules of this course will focus on how these various sorts of identifiers are used in cheminformatics applications. In this module, we are focusing on the communications tasks that almost all chemists engage in. A convenient mnemonic for these tasks is RSVP: Register, Search, View, Publish. Most forms of chemical representation were developed with these uses in mind. As we have touched on in previous modules, these activities are also evolving and may have different meanings to different disciplines. The demand for robust chemical representation increases as more and more chemical research data is published and re-purposed.
When representations originally built for these purposes are picked up for cheminformatics purposes, either directly or translated into more convenient formats, sometimes what had been features of these names and formulas in previous contexts turn into bugs. If you understand the original purposes served by these “bugs,” you’ll be better able to deal with them when they arise in your cheminformatics work.
2.1. Cheminformatics and structure representation
2.1.1. The what and why of cheminformatics
The term cheminformatics contains its own definition: it is the science of how we can make use of chemical information. Most often, data and information about chemical compounds is either directly about molecular structure (for example, a 2D structural formula, or 3D atomic coordinates for a particular conformation of a compound) or is tied to a molecular structure (for example, physical properties of a compound, which you identify by its structural formula).
Cheminformatics involves storing, finding, and analyzing these structures using the data-processing power of computers. In order for (human) chemists to rely on insights from cheminformatics, however, they have to be able to understand the methods that computer programs employ, the way in which computers store and analyze chemical structure, and the results that they produce.
Therefore, cheminformatics depends upon the use of representations of molecular structures and related data that are understandable to both human scientists and to machine learning algorithms.
The origins of the field trace back to library science. Librarians have been organising books full of words for centuries, but the notion of indexing, sorting, searching and retrieving information using molecular structures is more novel. Professionals whom we would now refer to as cheminformaticians have spent decades developing ways to handle molecular data in context: making sure scientists can find out what they need to know, and match chemical compounds with literature publications, measured properties, synthetic procedures, spectra, and computational studies.
More recently, the field of cheminformatics has been adopted by the pharmaceutical industry. Since the 1980s, it has been standard procedure for large drug companies to manage their collections of potential drug compounds using computer software. These days, cheminformatics is usually associated with drug discovery, since it is the pharmaceutical industry that provides the demand for software that can keep track of literally millions of unique molecules that are available for R&D purposes.
2.1.2 Communicating with chemists vs. communicating with computers
How does communication between chemists differ from communication between a chemist and a machine? If one chemist was to recommend to another that a reaction should be performed using "chloroform" as a solvent for a reaction, this would generally be a successful exercise in communication. For all practical purposes, this word is understood by every chemist, and has no ambiguity.
If this fact were to be communicated to a machine, things start to get a little murky. Humans are quite accustomed to learning common facts, and after sufficiently many years spent studying at university, they tend to become very good at looking up information and inferring missing data. Software algorithms, however, are supremely literal. Because "chloroform" is a so-called trivial name, there is no formula for converting it into the actual chemical structure that it represents, and so a machine will not be able to participate in this exchange of information unless it has been explicitly instructed as to what the word means, using a format that it can work with.
A more descriptive way to communicate the composition that is chloroform is by chemical formula, in this case CHCl3. If this were handed over to a computer program, it would be a simple matter for it to understand that the substance being described is a molecule with 5 atoms: 1 carbon, 1 hydrogen and 3 chlorine. Assembling this into a molecule with bonds is a very simple matter, because 4 of the atoms are normally monovalent, and one of them is normally tetravalent. It is quite simple to create a software algorithm that can join the atoms together in the most obvious way, which also happens to be correct.
Beyond such tiny simple molecules, difficulties soon arise. Some of these ambiguities affect human chemists in the same way that they affect machines. Consider the molecular formula of C3H6O, which is associated with multiple reasonable structures, including a ketone, an aldehyde, a cyclic alcohol, oxygenated alkenes and cyclic ethers, one of which exists as two enantiomers:
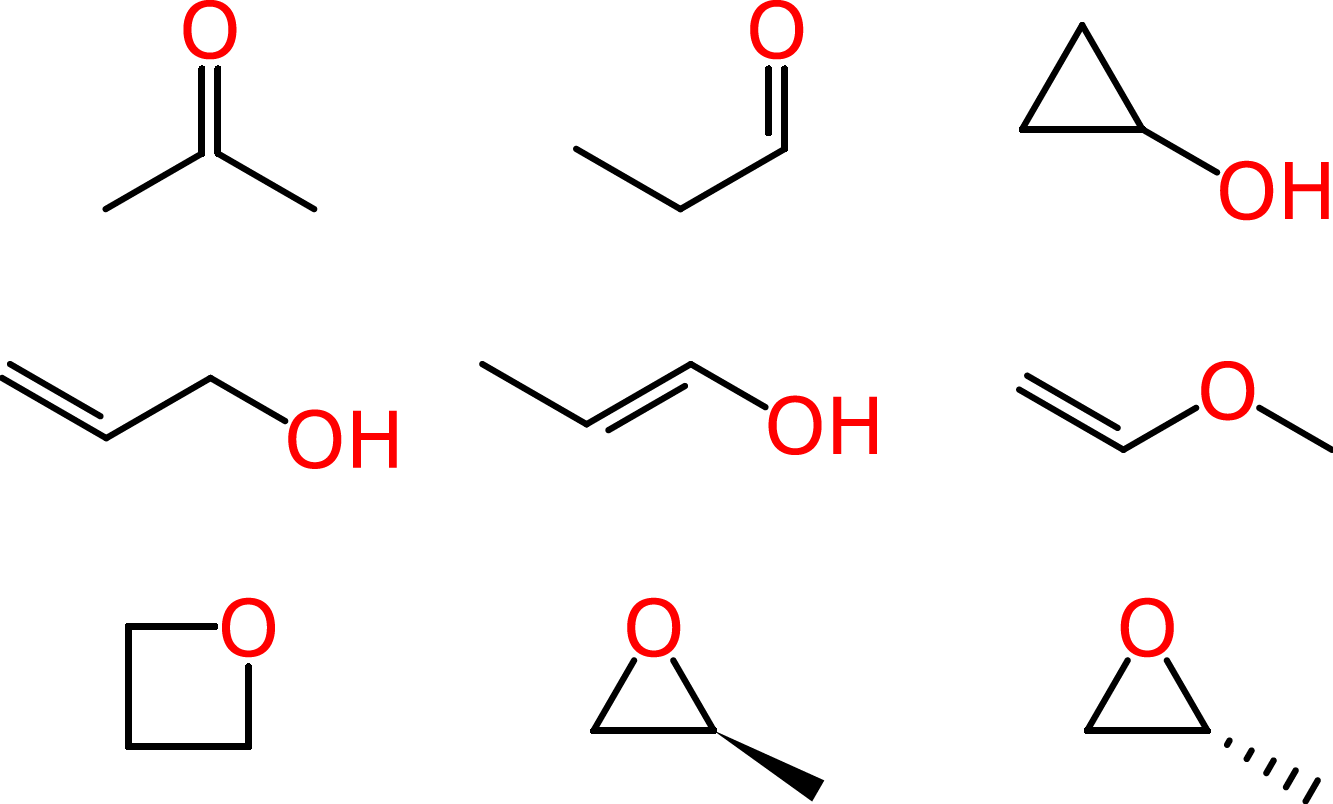
Since systematic IUPAC names are made according to formalized rules, they could, in principle, be used by both humans and computers. However, as we have seen in the previous unit, IUPAC names are often quite difficult for chemists to read, let alone to write. Even when chemists more or less follow IUPAC rules in naming their substances, they often take shortcuts, creating their own abbreviated notations and shortened words. These may be well-understood by two scientists working in the same laboratory or in the same field, but they might lead to misunderstanding when it comes time to communicate findings with the larger community.
Interacting with a machine is a form of communication. It has a lot in common with two scientists speaking to each other about their research, but it has its differences, too. Because cheminformatics is first and foremost about gathering scientific facts that originate from scientists, it is essential to find a way for the humans who do the chemistry to communicate using a vernacular that the machine can understand.
In this case it may be helpful to think of machines and cheminformatics software as an extremely pedantic bureaucrat with a set of rules that are utterly rigid with no flexibility or room for interpretation, working exclusively according to a formulaic script. In this metaphor, if you provide everything that the bureaucrat requires, in the right order, then you will be served perfectly, and be on your merry way. If not, then your task will fail: either the bureaucrat will prevent you from moving to the next step because the script forbids it, or your inputs will be erroneously accepted and moved onto the next level, and fail later on.
As with our bureaucrat, in cheminformatics, you’re dealing with a system governed by strict rules, and these rules are not too difficult to define. If you know the rules, then you can make the system work for you. If you don't know them, your experience will result in frustration and failure, and there is no guarantee that you will even find out what went wrong until much later.
2.1.3. Structural formulas as chemical graphs
As we discussed in Part 1 of this module, usually, the most effective way to communicate with another chemist about the structure of a compound is to draw its structural formula.
In order to do cheminformatics, we need to express chemical structure in a way that can be understood by machines as well as humans. It just so happens that structural formulas can be fairly directly mapped to a computer-friendly data structure. Structural formulas can be interpreted as a kind of graph: a set of nodes (in our case, atoms), certain pairs of which are linked by edges (in our case, bonds). For example, consider this structural formula for trifluoroacetic acid, CF3CO2H:
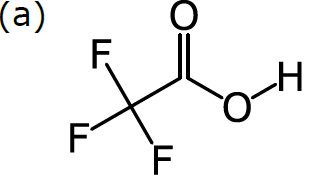
The diagram in (a) has no ambiguity, since all atoms are represented in a graph: there are 8 nodes, each of which is labelled according to the element, except for carbon (which is the default). Each of the 7 bonds is represented by an edge within the graph, and the bond order is represented by showing the number of lines. The way that this formula is drawn on paper is completely compatible with a data structure based on a labelled graph.
Such molecular graphs are typically stored in dedicated file formats designed for chemical information. (There are many to choose from; we’ll discuss the most popular ones a little later in this module and throughout the rest of this course.) This is good news, because cheminformaticians and computer scientists have come up with all kinds of clever data structures and algorithms for storing and analyzing datasets that can be represented as graphs.
However, just because structural formulas look like a graphs doesn’t mean that they always look like a chemically-meaningful graph – or like a graph with the same chemical meaning that the chemist who drew the structure intended. There is a long list of issues that need to be handled carefully in order to reliably encode all of the chemical information contained in a structural formula in a machine-readable manner. (Long enough to ensure that cheminformatics will continue to be a lively field of research for a very long time!)
Consider a more common way of drawing trifluoroacetic acid:
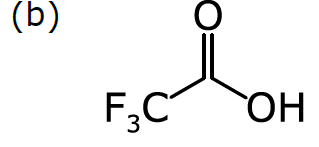
As we discussed in Part 1, chemists have become accustomed to condensing portions of structural formulas to make them easier to draw, read, and compare at a glance. The diagram (b) differs from the previous representation in that the carbon trisubstituted with fluoride has been collapsed into F3C, a single node (to use the terminology of graphs). So has the hydroxyl part of the acid (OH). A schematic form of the corresponding graph is shown in (c). In this case, the underlying graph has 4 nodes, not 8. The labels of these nodes are [O, C, F3C, OH]. Note that only two of these are elements from the periodic table. As shown, the structure represented by (c) is not a molecular graph, because some of its notes are labeled with something other than a symbol corresponding to an atom of a particular element.
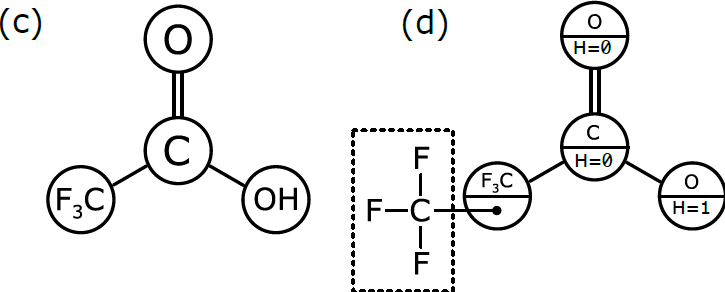
In order to be interpreted by a computer as if it were a molecule, the molecular graph needs to be labelled in a slightly different way, which is represented schematically in (d). Here, we have annotated the graph nodes in a more systematic way.
Note that each node now has two labels. Three of the nodes are labeled so that the primary property is an atomic element, and the secondary property is the number of hydrogen atoms attached to it. These three node definitions are [O:H=0, C:H=0, O:H=1].
The fourth node, which represents a more complex group of atoms, is labeled slightly differently. Its primary property is the label F3C, which is displayed for the benefit of human chemists reading the structural formula. However, its secondary property is labeled with another graph that stores the configuration of atoms and bonds that makes up F3C. This is the underlying chemical information that the human chemist picks up easily when she sees “F3C” in a structural formula, but that needs to be described explicitly in order for the computer to properly understand and use it.
Graph (d) is the best of both worlds. The structure can easily be displayed in the way that chemists expect to see it, but it can also be easily interpreted by a computer algorithm, because the definition follows rules that allow the full atomic structure to be reassembled behind the scenes.
Most importantly, always remember that just because a chemical structure has been digitized and stored on a computer does not mean that the information can be used by cheminformatics. In fact, most of the chemical structures that have been generated by scientists and put on computers are available not in a graph-based file format, but in a nonchemical graphics format. (There are two main types of such nonchemical image files: bitmapped and vector graphics. Neither is of much use for cheminformatics purposes.)
In order to make use of any of the capabilities that cheminformatics offers, the molecules involved must be represented as a molecular graph, rather than such generic print-ready forms. Furthermore, the molecular graph must be sufficiently well defined that an algorithm can use the information to piece together the complete structure. Every single atom and bond must be present and accounted for, in some way or another.
Different data structures do so in different ways, each of which has its advantages and disadvantages. For example, some approaches try to keep the representation as similar as possible to the human-friendly version (e.g. Kekulé forms for aromatic rings). Others favor a selection of properties that is more similar to the quantum wave-function of the molecule (e.g. use of resonance/aromatic bonds to denote equivalence).
2.1.4. Three dimensions of machine-readable chemical representation
A chemical structure representation contains two kinds of information: explicit and implicit. [H1] Explicit information is what’s directly represented in a data structure. Implicit information is what you (or a computer) can figure out from a data structure, given some knowledge of general principles and a little bit of work.
In general, data structures that contain less explicit information are more simple and compact, but they require more computation to draw chemical conclusions from them. Data structures that contain more explicit information take up more space and are at greater risk of containing inconsistencies, but they can be more quickly analyzed in a wider variety of ways.
In its essence, a molecular graph is a description of the relationships among a set of atoms that makes up a molecule. Like the condensed formulas and systematic names that we talked about in the first part of this module, a molecular graph can therefore be expressed in a one-dimensional (1D), or linear form. Such linear data structures are particularly well-suited to many of the best known cheminformatics applications, such as determining:
- whether molecules are the same.
- how similar they are, according to some metric.
- whether one molecular entity is a substructure of another.
- whether two molecules are related by a specific transformation.
- what happens when molecules are cut into pieces and grafted together at different positions.
In these and other applications of cheminformatics, linear representations have key advantages, especially when you’d like to handle huge numbers of structures very quickly (e.g. searching a large database).
However, chemists most frequently think about structure in 2D, and molecules actually exist in 3D physical space. Therefore, some data structures add 2D or 3D “diagram-like” coordinates for each atom.
In the 2D case, these 2D coordinates can be used to infer chemical information, such as the E/Z geometry of alkene-like double bond, or the cis/trans isomerism of ligands in a square planar metal complex, or substituents on a cyclic alkane. However, these coordinates are also descriptions for how exactly the structural formula is drawn. If you’re interested in communicating with other chemists as well as with a computer, you may wish to draw a structural formula in some particular way, to emphasize a certain bond or group. By storing the 2D coordinates of each atom, some data structures are designed to keep track of these choices, in order to facilitate human communication as well as machine analysis.
Other data structures are designed to represent the real 3D shape of a molecule. The molecular structure complete with a 3D (x,y,z) coordinate for each atom is often referred to as a conformation or a model. Such a data structure makes a scientific claim that the particular coordinates that it contains represent the molecule's actual shape, whether it be in solution, in a vacuum, or in the binding site of a protein. This opens up a whole new domain of computational chemistry, especially since most molecules have some flexibility. Even if a given conformation is the most stable, there often a number of competing shapes that must also be considered.
These coordinates may be determined experimentally (typically via x-ray crystallography). They may also be calculated, using force-fields (which treat atoms and bonds like "balls and springs" using classical physics), quantum chemistry (which solve the Schrödinger equation by various approximations), molecular dynamics (which model motion over time) or composite models such as docking (which are designed for specific environments, like peptide binding sites).
Keep in mind that even a data structure that provides 3D coordinates **may not tell you** where those coordinates come from. Knowing how a particular set of coordinates was determined is crucial to making intelligent use of it for cheminformatics purposes.
- Log in to post comments
2.2. Identifying chemical compounds on computers
2.2.1. How do they work?
Three forms of chemical notation have been developed especially for communication on computers and between humans and computers: connection tables, line notation and database record IDs.
2.2.1.1 Connection Tables
Molecular graphs are most often stored as a connection table. Connection tables do for computers what systematic nomenclature does for human chemists: they organize the structural information defined in a molecular graph in a form that is easier to read and to order in a list. The difference is that computers can read, sort, search, and group connection tables far faster than humans can work with systematic names or any other kind of formula or notation.
Connection tables may be generated in various ways and stored in different formats. However, you can think of a connection table, schematically, as a numbered list of entries corresponding to each of the atoms in a compound, where each entry indicates the identity of the atom, the other atoms to which it is linked by bonds, and the order of each of these bonds (single, double, triple).
The following depicts the connection table for benzoic acid in the MOL file format (with structural formula and annotations added):

No matter what form of input and output a computer program uses, anytime a computer does any analysis on chemical structure, it most likely makes use of connection tables. Connection tables are typically employed behind the scenes of chemical computer programs, out of the user’s view. As the table depicted above demonstrates, even when you can get a look at one, it’s pretty hard to learn much from it until the computer translates it into a more human-readable format.
2.2.1.2 Line Notation
Line notation is designed to express the structure of compounds in a form that is readable and writable (at least in principle) in a straightforward way by both humans and computers. The two most widely-used forms of line notation are InChI and SMILES. Both of these notations cover basic connectivity and topology of small organic molecules. InChI also has a condensed version of 27 characters called an InChIKey that can be used to search in Google and connect to database records that include this notation. You will learn more about InChI, InChIKey, and SMILES in Module 5.2.2.1.3 Database Record IDs
Databases that collect and organize information by chemical compounds will usually have a record ID system that identifies the profile of the compound as assembled in that database. These IDs can sometimes be considered de facto identifiers for the compounds themselves by the users of these databases. However, these ID systems are specific to their originating database organization and data structure and are not suitable to use in practice to identify compounds directly. Most record ID systems use non-chemically significant alpha-numeric strings and are highly unsuitable to function as proxies for molecular structure.
The most familiar system of chemical record IDs is the Chemical Abstracts Service Registry Number (CAS RN).
CAS RNs for many common compounds appear in many places online, including Wikipedia. However, most of these are unverified and for less well-known compounds, you must have access to SciFinder or another CAS system in order to easily obtain and use CAS RNs. The PubChem CID and the ChemSpider ID are two other record ID systems that are openly searchable. Human chemists can see and use these registry numbers, but on its own it tells you nothing about a compound (unless you happen to have memorized a particular compound’s registry number!).

|
SMILES |
O=C(O)C1=CC=CC=C1 |
|
InChI |
InChI=1S/C7H6O2/c8-7(9)6-4-2-1-3-5-6/h1-5H,(H,8,9) |
|
InChIKey |
WPYMKLBDIGXBTP-UHFFFAOYSA- N |
|
CAS RN |
65-85-0 |
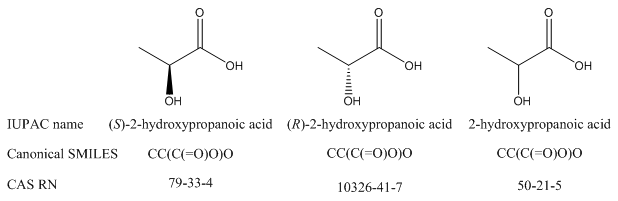
There is no way for a human reader to tell the relationship among compounds – even enantiomers – from registry numbers, other record IDs, or InChIKeys.
|
|
L-lactic acid (S enantiomer) |
D-lactic acid (R enantiomer) |
|
CAS RN |
79-33-4 |
10326-41-7 |
|
InChIKey |
JVTAAEKCZFNVCJ-REOHCLBHSA-N |
JVTAAEKCZFNVCJ-UWTATZPHSA-N |
2.2.2. Different purposes for chemical notation
As we’ve been discussing, two primary purposes for using chemical notation by both humans and computers are to communicate information about the molecular structure and to identify compounds. What information is required for each of these purposes depends on the needs of the user. What information is available to support these needs depends on the source notation. The various notation systems have properties related to uniqueness of a representation that impact their utility for these different purposes in different contexts.
Different chemical names and formulas serve different purposes. Some help you identify individual compounds. Others describe the structure of a compound very clearly, or help you sort and compare compounds according to their structure.
In an unambiguous system of notation, each name or formula refers to exactly one chemical entity, typically in a way that allows you to draw a structural formula for it. However, each chemical entity might be represented by more than one name or formula. This is true of IUPAC names and SMILES.
A canonical system of notation contains or generates a unique identifier for every chemical entity (a compound, a substructure, a ligand, a monomer, etc.) that can be represented within the system.
A canonical identifier may be the one and only representation for a chemical entity within a system (as with CAS Registry Numbers).
Alternatively, there may be several ways of representing a chemical entity using a certain system of nomenclature or notation, and an additional set of rules or an algorithm may be used to define one of these identifiers as canonical. This is true of Preferred IUPAC Names (PINs) and canonical SMILES (discussed in Module 5).
Ambiguous notation can refer to more than one chemical entity. This is true of most chemical names when used unsystematically (“octane,” used as a common term for all saturated hydrocarbons with eight carbon atoms rather than systematically to indicate the straight-chain isomer only). It is also true of empirical and molecular formulas.
In general, canonical notation is most reliable if your goal is to identify a compound within your application.
Unambiguous notation generally describes the structure of a compound effectively.
Ambiguous notation is often easier to interpret than canonical or unambiguous notation, and can be useful for sorting and comparing compounds.
Keep in mind: Just because a name or formula is canonical does not mean that it identifies a compound with absolute precision, especially outside the originating database. For example, there are three CAS registry numbers for lactic acid: one for each enantiomer and one for the racemic or unspecified version of the compound. You may need to use all three if you mean to refer to lactic acid in general. Similarly, as will be discussed in module 5, canonical SMILES does not take R/S stereoisomerism into account, so each enantiomer of a compound will have the same canonical SMILES formula.
2.2.3. Where did all of these names and formulas come from?
Names or formulas designed for one purpose can also be employed for another one. In fact, the ability to repurpose names and formulas in this way is part of what makes a particular kind of name or formula useful. However, a lot of the confusion that can arise over chemical names and notation arises from lack of awareness of the disjunction between the kinds of things that a name or formula was meant to do and the kind of things that you’re trying do with it.
This sort of re-use of notation happens a lot in cheminformatics – after all, some kinds of cheminformatics analysis weren’t even conceivable when most common forms of chemical names and formula first caught on. But the repurposing of notation isn’t unique to cheminformatics – in fact, as long as chemical names and formulas as we know them have been around, chemists have been re-using names, deciding that they fit other purposes better than the ones for which they were intended, or trying to change them in ways that undermine their original purpose.
For example, in 1892, a few dozen leading European chemists got together for the Geneva Nomenclature Congress, to develop for the first time an international system of organic chemical nomenclature. Some of them wanted unambiguous but non-canonical names, since it was often helpful, especially in teaching, to be able to name a compound in a way that emphasized one or another of its functional groups. They wished, for example, to be able to name vitamin C as either a lactone or a tetra-alcohol. Others thought that their nomenclature should be canonical as well as unambiguous. This would make it easier to use indexes of chemical substances. A reader would no longer have to try to think of all of the different names of a compound and check each of them, but would instead know that each compound could be found in an alphabetical list under one and only one name. At Geneva, the latter group triumphed, and the Congress created about sixty rules for translating structural formulas into canonical, unambiguous names.
Almost immediately, however, chemists started using these names in their teaching, research, and all sorts of other settings for which they weren’t designed. About the only place they weren’t used was in alphabetical indexes of chemical substances! The editors in charge of making these indexes found these canonical systematic names to be too difficult to write or to understand, and decided to incorporate a lot of trivial names into their indexes and/or to organize them according to molecular formulas rather than names.
Meanwhile, however, some chemists have become convinced that only the canonical systematic names that followed the Geneva nomenclature rules could guarantee clear, unambiguous communication. When another committee took up the question of international chemical nomenclature standards during the 1920s, it was the chemical editors who opposed canonical names, whereas chemists who were primarily interested in the use of names in teaching and laboratory research supported them. In the end, the result was the non-canonical IUPAC nomenclature rules, which offered numerous different options for systematically naming each compound, in the hope of satisfying as many of the different purposes for which chemists wished to use names as possible. Of course, one purpose that this approach could NOT satisfy was establishing a single identifier for each compound. That is why, over recent years, IUPAC has introduced even more rules for determining a canonical Preferred IUPAC Name for each compound. Both PINs and other changes in IUPAC nomenclature are also oriented toward making systematic names more easily readable by machines.

You don’t need to know any of the specifics of this history. What you do need to know is that chemists have been re-using notation and tinkering with how it works to make it fit the new use for a very long time. The lesson: carefully select the best existing kind of name, formula, or notation for your particular needs, be aware of the purpose for which it was originally designed, and think about whether you need to account for any differences between that purpose and yours.
- Log in to post comments
2.3. Translating and exchanging identifiers
2.3.1. From one chemical representation to another: translation and identifier exchange
We can summarize what we’ve learned so far:
- Different names and compounds may be designed to represent different sorts of chemical compounds, different structural features of these compounds, for different purposes.
- You can figure out what a particular name or formula DOES and DOES NOT tell you about the structure of a chemical entity by asking the kinds of questions that we have discussed above.
- Almost all chemical names and formulas, even ones designed for a very specific purpose, get re-used in other ways. Cheminformatics involves a lot of this sort of re-use.
Often, effective re-use of a particular name or formula involves swapping the identifier that you’ve found for another identifier for the same compound that’s more convenient for your purposes. For example, if you are interested in comparing the structures of a list of compounds for which you have registry numbers, you need to swap those registry numbers for structural formulas, connection tables, or another sort of representation that gives you the structural information you’re looking for.
The final section of this module provides an overview of how you should think about this process of swapping one kind of identifier for another.
There are two ways of exchanging identifiers: lookup and translation. In the case of lookup, you locate the identifier that you have in an existing database that lists various different identifiers for each compound, and you select the other identifier that you want. This is like using a thesaurus.
In the case of translation, you use a set of rules (or a computer uses an algorithm) to take apart one sort of representation of a compound and to create another sort of representation for the same compound.
Like words for the same object in different languages, even when two names or formulas are meant to refer to exactly the same compound, they differ in their connotations. They describe the compound’s structure in more or less specific ways, they emphasize different kinds of family relationships, and they draw upon different ways of understanding chemical objects and phenomena.
Scholars of literature like to emphasize that there is no such thing as a literal, perfect translation of a poem or novel from one language into another. Translation always involves decisions about what aspects of meaning to try to preserve and which to allow to become obscured. Literary language is often purposefully ambiguous, whereas chemical nomenclature and notation is extraordinarily precise. Nevertheless, even when it comes to chemical names and formulas, it is still often true that what one kind of chemical name or formula communicates cannot be perfectly expressed in another kind of name or formula.
Of course, this does not mean that translating one kind of name or formula into another is a bad idea. In fact, communication about chemical compounds depends upon chemists and computers constantly engaging in this kind of chemical translation. We will discuss how you can approach this process carefully, anticipate where misunderstandings might arise, and take measures to avoid them.
2.3.2. Validation
Naoki Sakai, a scholar of translation in literature and politics, has written, “Every translation calls for a countertranslation.” Any time you take an idea from one language A and put it into another B, you should think about how someone encountering the idea in the second language might translate it into the first.
This goes for chemical communication as well. When you “translate” a formula or name from one format to another, you should perform a countertranslation: that is, you should take your new name or formula and make sure that you can get back to the one you started with. The same goes for lookup: you should make sure that you can look up the identifier that you started with using the identifier that you generated.
This will help you be aware of what might have gotten lost or inadvertently added in translation. You won’t always be able to completely solve any potential problems that arise. Sometimes, the identifier that you started with and the one that you generate are not equally specific: for example, you can translate a structural formula into a single molecular formula, but you cannot translate that molecular formula back into a structural formula. As we have said, perfect translation is often not possible. But the validation exercises of countertranslation and reverse-lookup will help you be aware of any problems that might arise, so that you can figure out other ways to head them off.
Large chemical databases use validation and counter-translation as part of standardizing the data included in their chemical records. For example, they may collect data that includes both systematic names and molecular structures and run each of these name-to-structure and structure-to-name conversions to match any previous instances of these compounds in their databases or identify any potential errors
We’ll cover validation in greater detail later on in this course.
2.3.3. Provenance
Above, we discussed how the history of where a system of notation came from and the purpose for which it was designed affects what kinds of chemical information you can express using that sort of name or formula.
Individual names and formulas have their own individual histories, which we call their provenance. Where did you find the name or formula? Who put it there? Who or what created it, and why? Was it copied in from another system? Has it already been translated from another format? Can you trace it back to an original experiment, calculation, or hypothesis?
You should keep these questions in mind for your own sake. You should especially keep in mind that the person or computer that you’re communicating with won’t necessarily know the answers to these questions, and that this is a potential source of misunderstanding. They may also need to evaluate this information for their own subsequent re-use purposes.
Whenever you exchange one chemical name or formula for another via translation or lookup, you should keep track of:
- The source and the form of the original name or formula
- The tools and resources that you used in doing the lookup or (if applicable) the translation.
You may wish to share this information along with your new name or formula itself, in order to avoid miscommunication. Whether or not you do that, it’s your responsibility to keep track of your translation process in this way. Doing so will help you figure out what could have gone wrong when confusion arises (and perhaps to prove that the confusion isn’t your fault!)
2.3.4. Knowing your audience and user community
Chemical communication takes a wide variety of forms. Different formats of chemical nomenclature and notation are more appropriate for different settings. Sometimes, it’s pretty clear what format is right (or wrong) in a given situation. Systematic names aren’t usually much good in casual conversation; you can’t do a google search for a sketch of a structural formula; a computer can’t analyze a reaction mechanism using trivial names. However, there are plenty of cases in which it takes some thought to figure out what kind of name or formula is most effective for what you want to communicate. In addition to thinking about the object that you’re communicating about, you should always know with whom or with what you are communicating, and select an appropriate variety of name or formula.
One simple way to think about your audience is in terms of chemists and non-chemists, humans and computers.
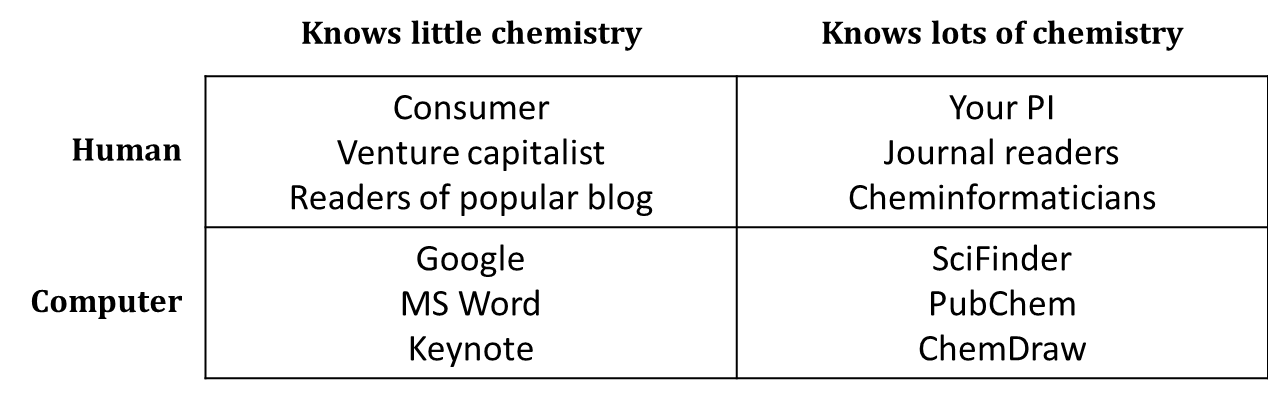
Of course, things are a little more complicated than this. Synthetic organic chemists have a certain area of expertise, and materials scientists another. Wikipedia “knows” a fair amount of chemistry because human experts in chemistry have manually added chemical information to many of its pages. Treat this simple table not as a set of boxes into which you must slot all of the different people and programs with which you communicate, but rather as a reminder that you should keep your audience in mind.
It is also useful to consider how translatable your chemical notation is for a diversity of unknown future cheminformatics applications. Follow common practices such as those used in the large public chemical databases, and/or carefully documenting your notation mapping and rules.
2.3.5. Further reading & references
Warr, W. A. Representation of chemical structures. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 557–579; DOI: 10.1002/wcms.36 (accessed May 29, 2104).
Warr, W. A. Some Trends in Chem(o)informatics. Chemoinformatics and computational chemical biology. Methods Mol. Biol. 2011, 672, 1–37; DOI: 10.1007/978-1-60761-839-3_1 (accessed May 29, 2104).
Wild, D. Introducing Cheminformatics: Navigating the world of chemical data. http://i571.wikispaces.com (accessed Sept. 29, 2015).
Willet, P. Chemoinformatics: a history. WIREs Comput. Mol. Sci. 2011, 1, 46–56; DOI: 10.1002/wcms.1 (accessed May 29, 2014).
- Log in to post comments
Module 4b: Assignment
2.4. Exercises (Module 4b)
2.4.1. Exercise 1 NOTE* You may want to download the MS Word Document at the top of the module homepage
Fill in the following table using both ChemSpider and PubChem and indicate where answers may differ. Use the new compound identity search in PubChem (click on the hexagon for structural formula, https://pubchem.ncbi.nlm.nih.gov/search/#collection=compounds&query_type=structure&query_subtype=identity)
|
molecular formula |
Structural formula |
Systematic name |
SMILES |
InChI |
CAS RN |
|
|
|
|
CC(=C)C=C |
|
|
|
|
|
|
|
|
|
|
|
|
Ammonium acetate |
|
|
|
|
C6H6O |
|
|
|
|
|
|
|
|
|
|
|
105-53-3 |
2.4.2. Exercise 2
Which of above form(s) of notation is/are preferable for:
- Ordering a specific compound from a supplier
- Identifying an important compound in a journal article
- Sorting a list of a bunch of compounds into chemically-meaningful groups
2.4.3. Exercise 3
Search PubChem for lactic acid (racemic) and its two enantiomers, shown in Module 4a, Figure III.a-e and in section 2.2.2 above.
- Paste the urls, IUPAC names, and CAS numbers that you find below.
- You should see something strange in your answer to Part b). What is it?
- Luckily, the makers of PubChem have followed some of the best practices that we’ve outlined above. What have they done that can help you get to the bottom of the inconsistency that you’ve discovered in parts (b-c)?
- Follow the clue that PubChem left you as to the source of the discrepancy and describe what might have happened with the data coming into Pubchem. (If you aren’t sure, what to do, click http://www.drugbank.ca/drugs/DB03066 and http://www.drugbank.ca/drugs/DB04398).
- Log in to post comments
Comments 2
Pubchem search
Chemical Structure Search vs. PubChem Search