2.2.1. How do they work?
Three forms of chemical notation have been developed especially for communication on computers and between humans and computers: connection tables, line notation and database record IDs.
2.2.1.1 Connection Tables
Molecular graphs are most often stored as a connection table. Connection tables do for computers what systematic nomenclature does for human chemists: they organize the structural information defined in a molecular graph in a form that is easier to read and to order in a list. The difference is that computers can read, sort, search, and group connection tables far faster than humans can work with systematic names or any other kind of formula or notation.
Connection tables may be generated in various ways and stored in different formats. However, you can think of a connection table, schematically, as a numbered list of entries corresponding to each of the atoms in a compound, where each entry indicates the identity of the atom, the other atoms to which it is linked by bonds, and the order of each of these bonds (single, double, triple).
The following depicts the connection table for benzoic acid in the MOL file format (with structural formula and annotations added):

No matter what form of input and output a computer program uses, anytime a computer does any analysis on chemical structure, it most likely makes use of connection tables. Connection tables are typically employed behind the scenes of chemical computer programs, out of the user’s view. As the table depicted above demonstrates, even when you can get a look at one, it’s pretty hard to learn much from it until the computer translates it into a more human-readable format.
2.2.1.2 Line Notation
Line notation is designed to express the structure of compounds in a form that is readable and writable (at least in principle) in a straightforward way by both humans and computers. The two most widely-used forms of line notation are InChI and SMILES. Both of these notations cover basic connectivity and topology of small organic molecules. InChI also has a condensed version of 27 characters called an InChIKey that can be used to search in Google and connect to database records that include this notation. You will learn more about InChI, InChIKey, and SMILES in Module 5.2.2.1.3 Database Record IDs
Databases that collect and organize information by chemical compounds will usually have a record ID system that identifies the profile of the compound as assembled in that database. These IDs can sometimes be considered de facto identifiers for the compounds themselves by the users of these databases. However, these ID systems are specific to their originating database organization and data structure and are not suitable to use in practice to identify compounds directly. Most record ID systems use non-chemically significant alpha-numeric strings and are highly unsuitable to function as proxies for molecular structure.
The most familiar system of chemical record IDs is the Chemical Abstracts Service Registry Number (CAS RN).
CAS RNs for many common compounds appear in many places online, including Wikipedia. However, most of these are unverified and for less well-known compounds, you must have access to SciFinder or another CAS system in order to easily obtain and use CAS RNs. The PubChem CID and the ChemSpider ID are two other record ID systems that are openly searchable. Human chemists can see and use these registry numbers, but on its own it tells you nothing about a compound (unless you happen to have memorized a particular compound’s registry number!).

|
SMILES |
O=C(O)C1=CC=CC=C1 |
|
InChI |
InChI=1S/C7H6O2/c8-7(9)6-4-2-1-3-5-6/h1-5H,(H,8,9) |
|
InChIKey |
WPYMKLBDIGXBTP-UHFFFAOYSA- N |
|
CAS RN |
65-85-0 |
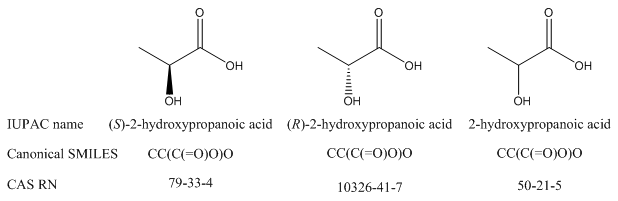
There is no way for a human reader to tell the relationship among compounds – even enantiomers – from registry numbers, other record IDs, or InChIKeys.
|
|
L-lactic acid (S enantiomer) |
D-lactic acid (R enantiomer) |
|
CAS RN |
79-33-4 |
10326-41-7 |
|
InChIKey |
JVTAAEKCZFNVCJ-REOHCLBHSA-N |
JVTAAEKCZFNVCJ-UWTATZPHSA-N |
2.2.2. Different purposes for chemical notation
As we’ve been discussing, two primary purposes for using chemical notation by both humans and computers are to communicate information about the molecular structure and to identify compounds. What information is required for each of these purposes depends on the needs of the user. What information is available to support these needs depends on the source notation. The various notation systems have properties related to uniqueness of a representation that impact their utility for these different purposes in different contexts.
Different chemical names and formulas serve different purposes. Some help you identify individual compounds. Others describe the structure of a compound very clearly, or help you sort and compare compounds according to their structure.
In an unambiguous system of notation, each name or formula refers to exactly one chemical entity, typically in a way that allows you to draw a structural formula for it. However, each chemical entity might be represented by more than one name or formula. This is true of IUPAC names and SMILES.
A canonical system of notation contains or generates a unique identifier for every chemical entity (a compound, a substructure, a ligand, a monomer, etc.) that can be represented within the system.
A canonical identifier may be the one and only representation for a chemical entity within a system (as with CAS Registry Numbers).
Alternatively, there may be several ways of representing a chemical entity using a certain system of nomenclature or notation, and an additional set of rules or an algorithm may be used to define one of these identifiers as canonical. This is true of Preferred IUPAC Names (PINs) and canonical SMILES (discussed in Module 5).
Ambiguous notation can refer to more than one chemical entity. This is true of most chemical names when used unsystematically (“octane,” used as a common term for all saturated hydrocarbons with eight carbon atoms rather than systematically to indicate the straight-chain isomer only). It is also true of empirical and molecular formulas.
In general, canonical notation is most reliable if your goal is to identify a compound within your application.
Unambiguous notation generally describes the structure of a compound effectively.
Ambiguous notation is often easier to interpret than canonical or unambiguous notation, and can be useful for sorting and comparing compounds.
Keep in mind: Just because a name or formula is canonical does not mean that it identifies a compound with absolute precision, especially outside the originating database. For example, there are three CAS registry numbers for lactic acid: one for each enantiomer and one for the racemic or unspecified version of the compound. You may need to use all three if you mean to refer to lactic acid in general. Similarly, as will be discussed in module 5, canonical SMILES does not take R/S stereoisomerism into account, so each enantiomer of a compound will have the same canonical SMILES formula.
2.2.3. Where did all of these names and formulas come from?
Names or formulas designed for one purpose can also be employed for another one. In fact, the ability to repurpose names and formulas in this way is part of what makes a particular kind of name or formula useful. However, a lot of the confusion that can arise over chemical names and notation arises from lack of awareness of the disjunction between the kinds of things that a name or formula was meant to do and the kind of things that you’re trying do with it.
This sort of re-use of notation happens a lot in cheminformatics – after all, some kinds of cheminformatics analysis weren’t even conceivable when most common forms of chemical names and formula first caught on. But the repurposing of notation isn’t unique to cheminformatics – in fact, as long as chemical names and formulas as we know them have been around, chemists have been re-using names, deciding that they fit other purposes better than the ones for which they were intended, or trying to change them in ways that undermine their original purpose.
For example, in 1892, a few dozen leading European chemists got together for the Geneva Nomenclature Congress, to develop for the first time an international system of organic chemical nomenclature. Some of them wanted unambiguous but non-canonical names, since it was often helpful, especially in teaching, to be able to name a compound in a way that emphasized one or another of its functional groups. They wished, for example, to be able to name vitamin C as either a lactone or a tetra-alcohol. Others thought that their nomenclature should be canonical as well as unambiguous. This would make it easier to use indexes of chemical substances. A reader would no longer have to try to think of all of the different names of a compound and check each of them, but would instead know that each compound could be found in an alphabetical list under one and only one name. At Geneva, the latter group triumphed, and the Congress created about sixty rules for translating structural formulas into canonical, unambiguous names.
Almost immediately, however, chemists started using these names in their teaching, research, and all sorts of other settings for which they weren’t designed. About the only place they weren’t used was in alphabetical indexes of chemical substances! The editors in charge of making these indexes found these canonical systematic names to be too difficult to write or to understand, and decided to incorporate a lot of trivial names into their indexes and/or to organize them according to molecular formulas rather than names.
Meanwhile, however, some chemists have become convinced that only the canonical systematic names that followed the Geneva nomenclature rules could guarantee clear, unambiguous communication. When another committee took up the question of international chemical nomenclature standards during the 1920s, it was the chemical editors who opposed canonical names, whereas chemists who were primarily interested in the use of names in teaching and laboratory research supported them. In the end, the result was the non-canonical IUPAC nomenclature rules, which offered numerous different options for systematically naming each compound, in the hope of satisfying as many of the different purposes for which chemists wished to use names as possible. Of course, one purpose that this approach could NOT satisfy was establishing a single identifier for each compound. That is why, over recent years, IUPAC has introduced even more rules for determining a canonical Preferred IUPAC Name for each compound. Both PINs and other changes in IUPAC nomenclature are also oriented toward making systematic names more easily readable by machines.

You don’t need to know any of the specifics of this history. What you do need to know is that chemists have been re-using notation and tinkering with how it works to make it fit the new use for a very long time. The lesson: carefully select the best existing kind of name, formula, or notation for your particular needs, be aware of the purpose for which it was originally designed, and think about whether you need to account for any differences between that purpose and yours.