Learning Objectives:
- Review publicly available chemical databases in different domains.
- Review text search, identity search, substructure/superstructure search, and similarity search.
- Review basic knowledge of molecular similarity methods.
- Perform chemical searches using the PubChem homepage and Chemical Structure Search page.
Module 6: Comparing and Searching Chemical Entities
1. Chemical Databases
These days many public online databases provide chemical information free of charge and the databases mentioned in this module are only a few examples of them. Note that these databases vary in size and scope.
- Chemical Databases
- PubChem: Chemical Information Respository at the US NIH
- ChemSpider: a Chemical Database Integrated with RSC'd Publishing Process
- ChEMBL: Literature-Extracted Biological Activity Information
- ChEBI: A Dictionary of Small Molecular Entity
- NIST Webbook: Thermodynamic and Spectroscopic Data of Chemicals
- DrugBank: Comprehensive Information on Drug Molecules
- HMDB: The Human Metabolome Database
- Toxnet: A Collection of Toxicological Information
- Protein Data Bank (PDB): A Key Source for Protein-Bound Ligand Structures
- Special Notes on Exchange/Integration
- Understanding Chemical Searches
1.9. Protein Data Bank (PDB): a key source for protein-bound ligand structures
The Protein Data Bank (PDB) is an archive of the experimentally determined 3-D structures of large biological molecules such as proteins and nucleic acids. These structures were determined primarily by using X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy. While PDB is not a small molecule database, it contains the 3-D structures of many proteins with small-molecule ligands bound to them. PDB allows users to search for proteins that an input small molecule binds to. Considering that it is not possible to experimentally determine how small molecules (such as drug or toxic chemicals) actually bind to their target proteins in a living organism, PDB is the most widely used resource for experimentally determined protein-bound structures of small molecules. The PDB are maintained by the Worldwide PDB (wwPDB)33, and freely accessible via the websites of its member organizations: PDBe (PDB in Europe)34,35, PDBj (PDB Japan)36,37, RCSB PDB (Research Collaboratory for Structural Bioinformatics PDB)38,39.
- Log in to post comments
1.10. Special notes on data exchange/integration
All the databases mentioned above are public databases that provide their contents free of charge, and in many cases, they also provide a way to download data in bulk and integrate them into one’s own database. Therefore, it is very common that different database groups exchange their information with each other. This often raises some technical concerns. For example, as mentioned in Module 5, different databases may use different chemical representations to refer to the same molecule. This may result in incorrect chemical structure matching between the databases, leading to incorrect data integration. In addition, when one database has incorrect information, this error often propagates into other databases.
- Log in to post comments
2. Understanding Chemical Searches
This section describes various searches that can be performed in PubChem. Currently PubChem has three different search interfaces:
- PubChem homepage (http://pubchem.ncbi.nlm.nih.gov)
- PubChem Chemical Structure Search (https://pubchem.ncbi.nlm.nih.gov/search/search.cgi)
- PubChem Search (https://pubchem.ncbi.nlm.nih.gov/search/).
The PubChem homepage provides a search interface for all three primary databases (e.g., Substance, Compound, and BioAssay). However, the search box on the PubChem homepage can accepts textual keywords only, and it is difficult to input non-textual queries (such as chemical structures). The PubChem Chemical Structure Search allows users to perform various searches using both textual and non-textual queries. This search interface is integrated with PubChem Sketcher, which enables users to provide the 2-D structure of a molecule as a query for chemical structure search. While the PubChem Chemical Structure Search is limited to chemical structure searches, the PubChem Search allows users to search for bioassays, bioactivities, patents, and targets as well as chemical structures, but it is still in beta testing. In this module, we use the PubChem homepage for name/text search and the Chemical Structure Search for others.
- Log in to post comments
2.1. Name/text search
Text search allows one to find chemical structures using one or more textual keywords, which may be chemical names (e.g., “aspirin”) or any word or phrase that describe molecules of interest (e.g., “cyclooxygenase inhibitors”). One can perform a text search from the PubChem homepage, by providing a text query in the search box. If the query is a phrase or a name with non-alphanumeric characters, double quotes should be used around the query. Various indices can be individually searched by suffixing a text query with an appropriate index enclosed by square brackets (for example, the query “N-(4-hydroxyphenyl)acetamide”[iupacname]). Numeric range searches of appropriate index fields can be performed using a “:” delimiter (for example, the query 100.5:200[molecularweight]for a molecular weight range search between 100.5 and 200.0 g/mol). One can see what search indices are available in PubChem from the drop-down menu on the “PubChem Compound Advanced Search Builder”, which can be accessed by clicking the “advanced” link (next to the “Go” button) on the PubChem homepage. Queries may be combined using the Boolean operators “AND”, “OR”, and “NOT”. These Boolean operators must be capitalized.
- Log in to post comments
2.2. Molecular formula search
Molecular formula search allows one to find molecules that contain a certain number and type of elements. Typically, molecular formula search returns by default molecules that exactly match the queried stoichiometry. For example, a query of “C6H6” will return all structures containing six carbon atoms, six hydrogen atoms and nothing else. However, molecular formula search implemented in some databases, including PubChem Chemical Structure Search, has an option to allow other elements in returned hits (e.g., C6H6O or C6H6N2O for the “C6H6” query).
- Log in to post comments
2.3. Identity search
Identity search is to locate a particular chemical structure that is “identical” to the query chemical structure. Although identity search seems conceptually straightforward, one should keep in mind that the word “identical” can have different notions. For example, if a molecule exists as multiple tautomeric forms in equilibrium, do you want to consider all these tautomers identical and search the database for all of them? If your query molecule has a chiral stereo center, should you consider both R- and S-forms in your search? In your identity search, do you want to include isotopically substituted species of the provided query molecule as well as the query itself? Depending on how to deal with these nuances of chemical structures, identical search will return different results. The identity search in the PubChem Chemical Structure Search allows users to choose a desired degree of “sameness” from several predefined options. To see these options, one need to expand the options section by clicking the “plus” button next to the “option” section heading.
- Log in to post comments
2.4. Substructure and superstructure search
When a chemical structure occurs as a part of a bigger chemical structure, the former is called a substructure and the latter is referred to as a superstructure. For example, ethanol is a substructure of acetic acid, and acetic acid is a superstructure of ethanol.
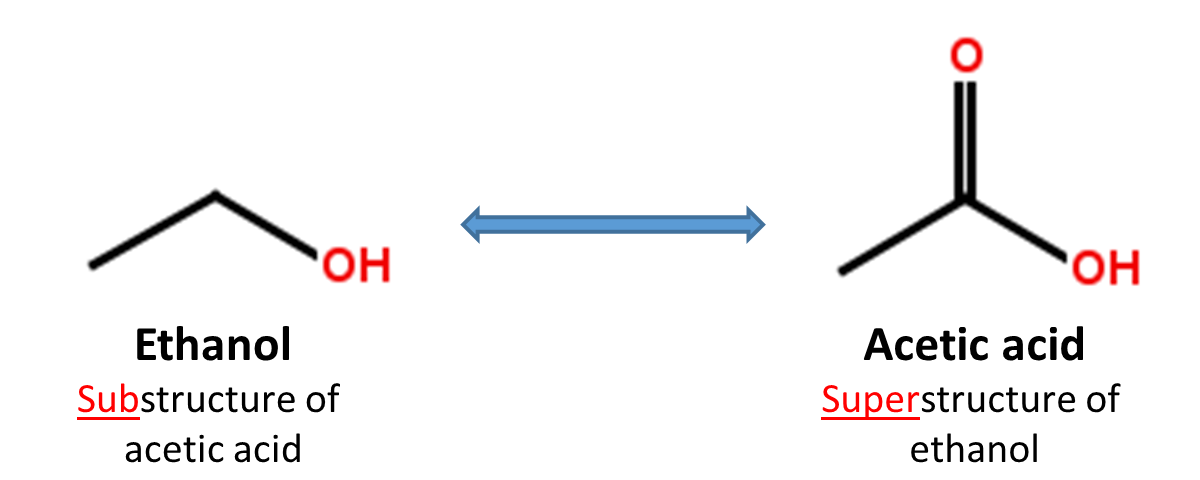
In substructure search, one provides an input substructure as a query to find molecules that contain the query substructure (that is, superstructures that contain the query substructure). On the contrary, superstructure search returns molecules that comprise or make up the provided chemical structure query (that is, substructures that is contained in the query superstructure). It should be noted that substructure search does not give you substructures of the query and that superstructure search does not return superstructures of the query.
It is possible to include explicit hydrogen atoms as part of the pattern being searched. For example, if you choose to do so, the SMILES queries [CH2][CH2][OH] and [CH3][CH][OH] will return molecules whose formula are R-CH2-CH2-OH and CH3-CH(R)-OH, respectively. Substructure/superstructure searches implemented in many databases remove by default explicit hydrogens from the query molecule prior to search, the two SMILES queries [CH2][CH2][OH] and [CH3][CH][OH] may give you the same result as what the SMILES query CCO does, unless you specify that explicit hydrogens should be included in pattern matching.
In addition to explicit hydrogen atoms, there are additional factors that may affect results of substructure/superstructure searches, for example, whether to ignore stereochemistry, isotopism, tautomerism, formal charge, and so on.
- Log in to post comments
2.5. Similarity search
Molecular similarity (also called chemical similarity or chemical structure similarity) is a fundamental concept in cheminformatics, playing an important role in computational methods for predicting properties of chemical compounds as well as designing chemicals with desired properties. The underlying assumption in these computational methods is that structurally similar molecules are likely to have similar biological and physicochemical properties (commonly called the similarity principle). Molecular similarity is a straightforward and easy-to-understand concept, but there is no absolute, mathematical definition of molecular similarity that everyone agrees on. As a result, there are a virtually infinite number of molecular similarity methods, which quantify molecular similarity. Similarity search uses a molecular similarity method to find molecules similar to the query structure.
2.5.1 Two-dimensional (2-D) similarity methods
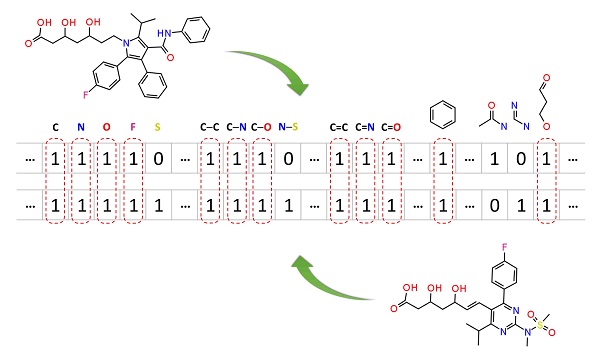
Molecular similarity methods can be broadly classified into two-dimensional (2-D) and three-dimensional (3-D) similarity methods. Typically, 2-D similarity methods use so-called molecular fingerprints, which encode structural information of a molecule into a binary string (that is, a string of 0’s and 1’s). The position of each number in this string corresponds to a particular fragment. If the molecule has a particular fragment, the corresponding bit position is set to 1, and otherwise to 0. Note that there are many different ways to design molecular fingerprints, depending on what fragments are included in the fingerprint definition. PubChem uses its own fingerprint called PubChem subgraph fingerprints.
In 2-D similarity methods, structural similarity between two molecules is estimated by comparing their molecular fingerprints. Their similarity is quantified as a so-called similarity score or similarity coefficient. While several different methods can be used for computation of a similarity score, the underlying ideas are the same as each other: if the two fingerprints have 1’s at the same position, it means that both compounds have the same fragment, and if the molecules share more common fragments, they are considered to be more similar. In conjunction with the PubChem subgraph fingerprints, PubChem 2-D similarity method use the Tanimoto coefficient
![]()
where NA and NB are the number of bits set in the fingerprints for molecules A and B, respectively, and NAB is the number of bits set in both fingerprints. The Tanimoto score ranges from 0 (for no similarity) to 1 (for identical molecules). 2-D Similarity search returns molecules whose similarity scores with the query molecule are greater than or equal to a given Tanimoto cut-off value.
- Log in to post comments
2.5.2. PubChem 3-D similarity method
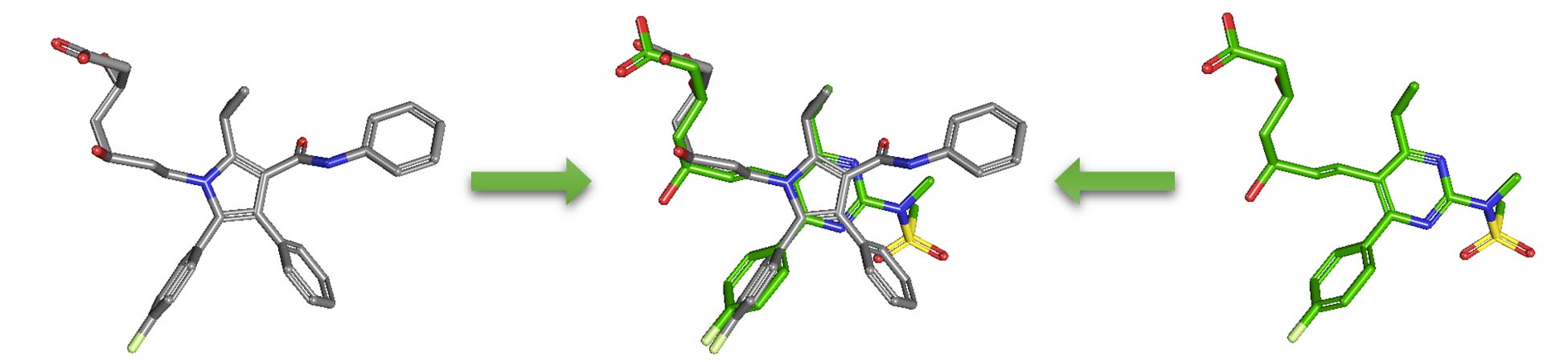
As an alternative to 2-D similarity search, 3-D similarity search can also be performed using the “3D conformer” tab in PubChem Chemical Structure Search. 3-D similarity methods use the 3-D structures (that is, conformations) of molecules. PubChem’s 3-D similarity method is based on the atom-centered Gaussian-shape comparison method by Grant, Gallardo and Pickup, implemented in the Rapid Overlay of Chemical Structures (ROCS). While the underlying mathematics of this approach is beyond the scope of this module, what this method essentially does is to find the "best" alignment of the 3-D structures of two molecules, which gives the maximized overlap between them. The 3-D similarity method quantifies the 3-D molecular similarity using three metrics.
- Shape-Tanimoto (ST): quantifies steric shape similarity between two conformers.
- Color-Tanimoto (CT): quantifies the overlap of functional groups between two conformers, such as hydrogen bond donors and acceptors, cations, anions, rings, and hydrophobes.
- Combo-Tanimoto (ComboT): the sum of ST and CT scores between two conformers. It takes into account the shape similarity (ST) and functional group similarity (CT) simultaneously.
Because both the ST and CT scores range from 0 (for no similarity) to 1 (for identical molecules), the ComboT score may have a value from 0 to 2 (without normalization to unity). Note that the ST, CT and ComboT scores between two molecules can be evaluated in two different molecular superpositions: (1) in the ST- or shape-optimized superpositions, and (2) in the CT- or feature-optimization superpositions. In the ST-optimization approach, the shape overlap between the molecules (that is, the ST score) is maximized and the single-point CT score is evaluated at that superposition. On the contrary, the CT-optimization considers both ST and CT scores to find the best superposition between molecules, and the single-point ST score is computed at that superposition.
- Log in to post comments
References
(6) ChemSpider (http://www.chemspider.com) (Accessed on 6/29/2015).
(7) Pence, H. E.; Williams, A. J. Chem. Educ. 2010, 87, 1123.
(8) ChemSpider SyntheticPages (CSSP) (http://cssp.chemspider.com/) (Accessed on 7/13/2015).
(9) ChEMBL (https://www.ebi.ac.uk/chembl/) (Accessed on 7/10/2015).
(11) Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations (http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm) (Accessed on 7/13/2015).
(12) DailyMed (http://dailymed.nlm.nih.gov/) (Accessed on 7/13/2015).
(13) ChEBI (https://www.ebi.ac.uk/chebi/) (Accessed on 6/29/2015).
(15) NIST Chemistry Webbook (http://webbook.nist.gov/chemistry/) (Accessed on 6/29/2015).
(16) Linstrom, P. J.; Mallard, W. G. J. Chem. Eng. Data 2001, 46, 1059.
(17) DrugBank (http://www.drugbank.ca/) (Accessed on 7/10/2015).
(18) About DrugBank (http://www.drugbank.ca/about) (Accessed on 7/13/2015).
(20) The Human Metabolome Database (HMDB) (http://www.hmdb.ca/) (Accessed on 7/10/2015).
(21) About the Human Metabolome Database (HMDB) (http://www.hmdb.ca/about) (Accessed on 7/13/2015).
(23) ToxNet (http://toxnet.nlm.nih.gov/) (Accessed on 7/9/2015).
(24) Factsheet - Toxicology Data Network (TOXNET) (http://www.nlm.nih.gov/pubs/factsheets/toxnetfs.html) (Accessed on 7/9/2015).
(25) Wexler, P. Toxicology 2001, 157, 3.
(26) Fowler, S.; Schnall, J. G. Am. J. Nurs. 2014, 114, 61.
(27) ChemIDplus (http://chem.sis.nlm.nih.gov/chemidplus/chemidlite.jsp) (Accessed on 7/9/2015).
(28) Fact Sheet - ChemIDplus (http://www.nlm.nih.gov/pubs/factsheets/chemidplusfs.html) (Accessed on 7/9/2015).
(29) Hazardous Substances Data Bank (HSDB) (http://toxnet.nlm.nih.gov/newtoxnet/hsdb.htm) (Accessed on 7/9/2015).
(30) Fact Sheet - Hazardous Substances Data Bank (HSDB) (http://www.nlm.nih.gov/pubs/factsheets/hsdbfs.html) (Accessed on 7/9/2015).
(31) Comparative Toxicogenomics Database (CTD) (http://toxnet.nlm.nih.gov/newtoxnet/ctd.htm) (Accessed on 7/9/2015).
(32) Fact Sheet - Comparative Toxicogenomics Database (CTD) (http://www.nlm.nih.gov/pubs/factsheets/ctdfs.html) (Accessed on 7/9/2015).
(33) Worldwide Protein Data Bank (wwPDB) (http://www.wwpdb.org/) (Accessed on 7/9/2015).
(34) Protein Data Bank in Europe (PDBe) (http://www.ebi.ac.uk/pdbe/) (Accessed on 7/9/2015).
(36) Protein Data Bank Japan (PDBj) (http://pdbj.org/) (Accessed on 7/9/2015).
(38) RCSB Protein Data Bank (RCSB PDB) (http://www.rcsb.org/pdb/) (Accessed on 7/9/2015).
Module 6: Questions
Questions
- Conceptually, data in a database are stored in the same way as we would record them in a table or excel spreadsheet. The rows in the table correspond to compounds, and the columns correspond to properties or descriptions for those compounds (e.g., melting and boiling points, chemical names, toxicity, bioactivity, target proteins, and so on). These columns are commonly called “data fields”. You may want to perform a search against all data fields or only a particular field. To search the chemical name field of the records in the PubChem Compound database, a chemical name query needs to be suffixed with either of the “[synonym]” or “[completesynonym]” index. The “[synonym]” index will search for molecules whose names contain the query chemical name as a part (that is, partial matching), and the “[completesynonym]” index will search for those whose names completely match the query (that is, exact matching). If no index is given after the query, PubChem will search all data fields.
Go to the PubChem homepage (https://pubchem.ncbi.nlm.nih.gov) and select the “Compound” tab above the search box. Provide the following queries in the search box and click the “Go” button. How many hits do you get for each search? Clicking the image of each compound will direct you to the Compound Summary page of that compound, which provides comprehensive information on the compound. On the Compound Summary page of each compound, check the “Depositor-Supplied Synonyms” section to see if any of the chemical names of the molecule contains the string “zyrtec”.
(1) zyrtec
(2) zyrtec[synonym]
(3) zyrtec[completesynonym]
- Usinf this Link, To perform an identity search for Cymbalta (CID 60835), go to the Chemical Structure Search page (https://pubchem.ncbi.nlm.nih.gov/search/search.cgi) and select the “Identity/Similarity” tab. Expand the “Options” section by clicking the “plus” button and select the “Identical Structures” with “same connectivity” from the drop-down menus. Expand the Filters section and limit the number of covalent units to 1 (by setting the range to “from 1 to 1”). Provide the query CID in the search box and run the search. Repeat the search with the “same isotopical labels” option selected. Explain how the two different options affect the identity search results.
- Perform a 2-D similarity search using CID 5090 as a query. Select the “Identity/Similarity” tab and expand the Options sections by clicking the “plus” button next to the “Options” section heading. Select the “Similar Structures” and “95%” from the drop-down menus. Expand the Filters section and limit the number of covalent units to 1. Provide the CID query in the search box and press the “search” button. Repeat the search with the following similarity search threshold: 90%, 85%, and 80%. How many records are returned for each search?
The right column of the last search result page (for threshold >= 80%) shows what kind of information is available for the returned compounds. Click the “Pharmacological Actions” link under “BioMedical Annotation” to choose the compounds with the Pharmacological Action annotations. For each compound, check the information under the “Pharmacology and Biochemistry” section. What pharmacological actions do these compouns have?
- Select the “3D Conformer” tab to perform a 3-D similarity search using CID 5090 as a query. Expand the Options section and select the “(Sort results by) Shape-then-feature” and “(output to) NCBI Entrez” options from the drop-down menus. Expand the Filters section and limit the covalent unit count to 1. Type the query CID in the search box and press the “search” button. How many compounds are returned? How many CIDs have pharmacological action annotations. Compare the results from 3-D similarity search with those from 2-D similarity search.
- Log in to post comments