Learning Objectives
- Know how to formulate a PUG-REST request URL.
- Know how to access PubChem data from a spread sheet (in Google Sheet)
- Know how to access PubChem data from a python script.
Table of Contents
1. Useful resources for students with no programming background
In this module, we will learn how to “programmatically” access PubChem and other public databases. Cheminformaticians often write computer programs to process a large amount of chemical data and to automate some tasks that are routinely performed. Therefore, computer programming is an essential skill for future cheminformaticians. While this module does not require prior knowledge of computer programming, it is highly recommended that students with no or little programming background build some basic computer programming skills. There are many online resources that help you learn computer programming and below are some of them.
- w3schools.com (https://www.w3schools.com)
- Tutorialspoint.com (https://www.tutorialspoint.com/index.htm)
- Code.org (https://code.org)
- Code Academy (https://www.codecademy.com)
- Exercism (http://exercism.io)
- Cloud9 (https://c9.io)
- Ideone (https://ideone.com)
- LearnPython.org (https://www.learnpython.org)
- Python Challenge (http://www.pythonchallenge.com)
In addition, an introductory material about Web APIs (Application Programming Interfaces) is available at the OLCC web site (as a Special Topic Module).
- Programmatic Access to Data (by Jordi Cuadros)
(http://olcc.ccce.divched.org/Spring2017OLCCSpecialTopic-Programmatic%20Access%20to%20Data)
The present module focuses on accessing PubChem data, but many other public chemical information resources also provide programmatic access to their data. Some examples complied by the OLCC Cheminformatics Faculty are available at this URL:
- Programmatic Access to Web-Based Chemical Information – Examples
(http://olcc.ccce.divched.org/Spring2017OLCCModule7TLO1)
2. Overview of Programmatic Access to PubChem
Currently PubChem1-3 contains more than 235 million depositor-provided substance descriptions, 94 million unique chemical structures and 232 million biological test results from one million assays, covering more than 10 thousand unique protein target sequences. Many researchers in the biomedical science community have a great interest in programmatic access to this vast amount of data because it presents new opportunities for data-driven research in a “big data” era. PubChem provides several ways for programmatic access to its data, including:
- Entrez Utilities (also called E-Utilities or E-Utils)
- Power User Gateway (PUG)
- PUG-SOAP
- PUG-REST
E-Utilities, used for programmatic access to information contained in the Entrez system, are suited for accessing text or numeric-fielded data, they cannot deal with more complex types of data specific to PubChem, such as chemical structures and tabular bioactivity data. Thus, PubChem provides additional programmatic access routes specialized for PubChem data and analysis services: PUG, PUG-SOAP, and PUG-REST. While suitable for low-level programmatic access to PubChem, PUG exchanges data through a complex eXtended Markup Langauge (XML) schema that requires some expertise to use. For the sake of user-friendliness and for integration with a variety of third party tools, PubChem provides two easier-to-use web service access methods: PUG-SOAP, which uses the simple object access protocol (SOAP) and PUG-REST, which is a Representational State Transfer (REST)-style interface. An overview of these programmatic access routes to PubChem is given in the following paper:4
- PUG-SOAP and PUG-REST: Web Services for Programmatic Access to Chemical Information in PubChem
Kim et al., Nucleic Acids Res. 2015, 43(W1), W605-W611.
(http://dx.doi.org/10.1093/nar/gkv396).
In this module, we will learn how to access PubChem using PUG-REST, because it is the simplest to use and learn. More in-depth information on programmatic access to PubChem is described in these documents:
- PUG-REST
- PUG-REST Help (http://pubchem.ncbi.nlm.nih.gov/pug_rest/PUG_REST.html)
- PUG-REST Tutorial (http://pubchem.ncbi.nlm.nih.gov/pug_rest/PUG_REST_Tutorial.html)
- Programmatic Retrieval of Small Molecule Information from PubChem Using PUG-REST (in press).
[Available to logged-in students at bottom of this module.]
- E-Utilities
- Entrez Programming Utilities Help (http://www.ncbi.nlm.nih.gov/books/NBK25501/)
- E-Utilities Quick Start (http://www.ncbi.nlm.nih.gov/books/NBK25500/)
- PUG
- PUG-SOAP
- PUG-SOAP Help (http://pubchem.ncbi.nlm.nih.gov/pug_soap/pug_soap_help.html
- PUG-SOAP Client Help (http://pubchem.ncbi.nlm.nih.gov/pug_soap/client_help.html)
- PUG-SOAP Web Service Reference (http://pubchem.ncbi.nlm.nih.gov/pug_soap/PUG_SOAP.html)
3. PUG-REST
3.1. Concepts and Syntax of PUG-REST requests
PUG-REST is the simplest to use and learn among the existing programmatic access methods to PubChem. Importantly, because information necessary for a PUG-REST request can be encoded into a single Uniform Resource Locator (URL) that can be written by hand without programming expertise. Conceptually, a web service request from the user to PubChem requires three pieces of information:
- input: a list of PubChem identifiers of interest (e.g., CID, AID, SID).
- operation: what to do with the input identifiers.
- output: the format of the output from the operation.
In PUG-REST, these three pieces of information are encoded into an URL in the following format:
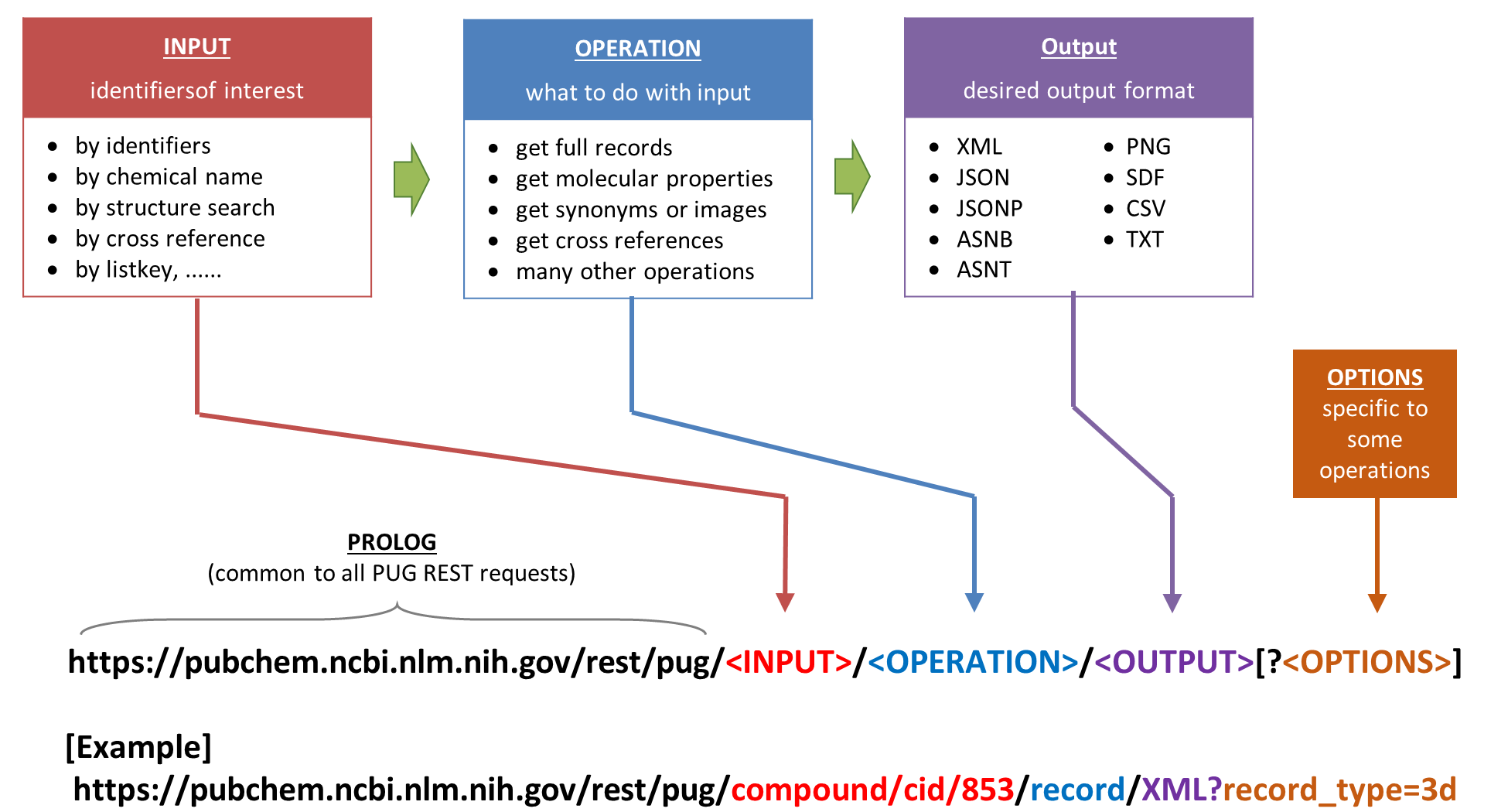
Some tasks require additional pieces of information that do not fit into the three-part PUG-REST URL. They should be provided as a list of ‘&’-separated option name and option value pairs, following the question mark (“?”) appended at the end of the request URL. Some examples are presented in next section, but there are much more things that users can do through PUG-REST. To get more detailed information on PUG-REST, read the following four articles:
- PUG-SOAP and PUG-REST: Web Services for Programmatic Access to Chemical Information in PubChem
Kim et al., Nucleic Acids Res. 2015, 43(W1), W605-W611.
(http://dx.doi.org/10.1093/nar/gkv396). - Programmatic Retrieval of Small Molecule Information from PubChem Using PUG-REST (in press).
[Available to logged-in students at bottom of this module.] - PUG-REST Help (http://pubchem.ncbi.nlm.nih.gov/pug_rest/PUG_REST.html)
- PUG-REST Tutorial (http://pubchem.ncbi.nlm.nih.gov/pug_rest/PUG_REST_Tutorial.html)
3.2. PUG-REST examples
-
Getting the full record of a compound record.
One of the most common tasks requested through PUG-REST is to retrieve all computed properties for a given chemical or chemicals. The following example URLs retrieve the full record of acetone in an XML format.
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/180/record/XML
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/acetone/record/XML
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/inchikey/CSCPPACGZOOCGX-UHFFFAOYSA-N/record/XML
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/inchikey/CSCPPACGZOOCGX-UHFFFAOYSA-N/record/XML?record_type=2d
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/inchikey/CSCPPACGZOOCGX-UHFFFAOYSA-N/record/XML?record_type=3d
In the above examples, the input compound is specified by CID (CID 180), name (acetone), and InChiKey (CSCPPACGZOOCGX-UHFFFAOYSA-N). The input identifiers can also be specified by SMILES or InChI strings, although special care needs to be taken because these identifiers contain special characters (such as “/”) that cause conflicts with the URL syntax.4 In addition, the hits returned from a structure search (e.g., identity/similarity search or sub/superstructure search) can be used as the input identifiers for a PUG-REST request. [It is highly recommended to use the synchronous “fast” inputs for structure searches (e.g., fastidentity, fastsimilarity_2d, fastsimilarity_3d, fastsubstructure, fastsuperstructure, and fastformula). Read the “Asynchronous operations in PUG-REST” section of this paper.]
The keyword “record”, followed by the input identifier, invokes the full record retrieval of the input compound. By default, the “record_type” option is set to “2d”, meaning that the information derived from the 2-D structure of the input compound will be returned. By setting this option to “3d”, one can get the information derived from the 3-D structure. Therefore, examples (a) through (d) returns 2-D information, and example (e) returns 3-D information.
-
Getting the 2-D and 3-D structure image of a compound
Below are examples of the molecular structure image retrieval through PUG-REST.
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/180/record/PNG
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/180/record/PNG?record_type=2d
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/180/record/PNG?record_type=3d
When the output file format for full record retrieval is set to “PNG”, the image of the input compound is retrieved. The 3-D image of the input compound can be retrieved by setting the “record_type” option to “3d”. When a list of compounds are specified as the input, the image of only the first compound on the list will be returned.
-
Getting molecular properties of a set of compounds
One may download molecular properties for a set of compounds, as in the following example:
In this example, the molecular formula, molecular weight, canonical SMILES, Heavy Atom Count, and XLOGP value for 4 compounds are retrieved in a CSV format, which can be read in a spread sheet program like Excel and Google Sheet. A list of the molecular properties available through PUG-REST can be found in the PUG-REST specification document:
https://pubchem.ncbi.nlm.nih.gov/pug_rest/PUG_REST.html#_Toc458584223
-
Getting a list of CIDs whose synonym is or contains “atorvastatin”
Through PUG-REST, one can perform a search by synonym.
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/atorvastatin/cids/txt
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/atorvastatin/cids/txt?name_type=complete
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/name/atorvastatin/cids/txt?name_type=word
By default, the “name_type” option is set to “complete”, meaning that only those whose synonym completely matches the input chemical name will be returned. One can perform partial synonym matching, by setting this option to “word”. These search options are conceptually equivalent to an Entrez search with Entrez indices “[completesynonym]” and “[synonym]”.
-
Getting a list of CIDs for compounds identical to a query compound
Below are examples of PUG-REST request URLs for identity search.
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastidentity/cid/4594/cids/TXT
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastidentity/cid/4594/cids/TXT?identity_type=same_stereo_isotope
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastidentity/cid/4594/cids/TXT?identity_type=same_connectivity
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastidentity/cid/4594/cids/TXT?identity_type=same_isotope
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastidentity/cid/4594/cids/TXT?identity_type=same_stereo
Note that various “contexts” of identity can be selected using the “identity_type” option. By default, this option is set to “same_stereo_isotope”, which returns compounds identical to the query compound in both stereochemistry and isotopism. When it is set to “same_connectivity”, the identity search will returns molecules with the same connectivity (ignoring stereochemistry and isotopism).
-
Getting a list of CIDs for compounds with a given substructure
The following examples perform substructure searches through PUG-REST.
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastsubstructure/cid/6857523/cids/TXT
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastsubstructure/cid/6857523/cids/TXT?StripHydrogen=false
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastsubstructure/cid/6857523/cids/TXT?StripHydrogen=true
By default, the substructure search will keep explicit hydrogen atoms in the query substructure. By setting the StripHydrogen parameter to “true”, one can strip off hydrogen atoms from the query substructure before performing a substructure search.
-
Getting a list of CIDs for compounds with a given molecular formula
The following examples show how to perform a molecular formula search through PUG-REST.
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastformula/C6H12O6/cids/TXT
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/fastformula/C6H12O6/cids/TXT?AllowOtherElements=true
By default, the search results from a molecular formula search will exactly match the entered stoichiometry. One may allow other elements in the returned results by using the “AllowOtherElements=true” option.
-
Getting a list of AIDs for assays that target a given protein.
One can retrieve assays that are performed against a particular target, which can be specified by gene symbol, NCBI’s Gene ID, or NCBI’s global identifier (gi) (for protein sequences).
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/target/genesymbol/HMGCR/aids/TXT
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/target/geneid/3156/aids/TXT
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/target/gi/118573791/aids/TXT
-
Getting a list of compounds tested in an assay.
The following examples show how to get a list of compounds tested in a given assay.
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/aid/1053202/cids/XML
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/aid/1053202/cids/XML?cids_type=all
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/aid/1053202/cids/XML?cids_type=active
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/aid/1053202/cids/XML?cids_type=inactive
Using the “cids_type” option, one can download all compounds tested in an assay or only those compounds tested to be active (or inactive).
-
Getting bioactivity data determined in an assay.
It is possible to download the bioactivity data for a given assay through PUG-REST.
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/aid/1053202/record/CSV
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/aid/1053202/record/CSV?cid=823611,1270560,2104765
It is also possible to download the bioactivity data for a particular compound or compounds tested in the input assay by providing the comma-separated list of CIDs of interest after the question mark.
-
Getting a list of assays in which a compound was tested.
Through PUG-REST, one can get a list of assays in which a given compound was tested. Below are some examples.
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/5329102/aids/XML
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/5329102/aids/XML?aids_type=all
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/5329102/aids/XML?aids_type=active
- https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/5329102/aids/XML?aids_type=inactive
By adjusting the “aids_type” parameter to “active” (or “inactive), one can retrieve assays in which the compound is tested to be active (or inactive). By default, any assays in which the input compound is tested are retrieved.
3.3. Accessing PubChem data from a spread sheet program
Through PUG-REST, one can access PubChem data from a spread sheet software, such as Google Sheet or MicroSoft Excel. Below is an example Google Sheet file that shows how to auto-populate data from PubChem into the spread sheet.
https://docs.google.com/spreadsheets/d/12-UkxgCxNVp2Ceim47TRffhqquLi2TFPKj3PJpii7bY/edit?usp=sharing
In this example, the function IMAGE() is used to retrieve the molecular images in the PNG format, and IMPORTXML() is used to get the record name and some molecular properties. While the IMPORTXML() function is used to get data in an XML format, the IMPORTDATA() function should be used to get PubChem data in CSV or TXT format. Note that the PUG-REST request URL used in the IMPORTXML() contains a comma-separated list of the input CIDs to retrieve data for all CIDs at a single request. While it is possible to make ten PUG-REST requests (one for each CID) to get the same data, it is highly recommended that users should minimize the number of requests. On the other hand, the image retrieval through PUG-REST supports only one input CID at a time, and therefore ten PUG-REST requests are made to get the images for all CIDs.
It is also possible to use MicroSoft Excel to do the same task as done in the above Google Sheet example. The WEBSERVICE() function in MicroSoft Excel 2013/2016 is equivalent to IMPORTXML() and IMPORTDATA() in Google Sheet. It returns the data retrieved from a web service request (in both XML and CSV/TXT). However, MicroSoft Excel does not have a built-in function equivalent to IMAGE() in Google Sheet, one needs to write a script for the image retrieval in VBA (Visual Basic for Applications), which is beyond the scope of this module.
3.4. Accessing PubChem data from a script
This section introduces how to make a PUG-REST request from a script, with example Python scripts. These examples are simple and short enough for students with no programming background to understand. You can modify and run these scripts on your web browser. While the examples are written in Python, it is possible to do the same tasks using other programming languages.
Example 1: Search by chemical name (https://trinket.io/library/trinkets/f009cf46ee)
This example searches PubChem for compounds whose name is “atorvastatin” and retrieve their full record. All lines beginning with the “#” characters are comments, which are ignored during the execution of a script. These comments are usually used to provide human-readable explanations about the script.
Without the comments and blank lines, only the remaining three lines are actually executed. In line 8, the “urllib.request” module is imported, which contains the definition of the function “urllib.request.urlopen()” in line 11. This function makes a web service request to the URL provided within the parentheses. The returned result is stored in the “request” object. In line 14, the returned data are read from the object and printed out.
Example 2: Molecular property retrieval (https://trinket.io/library/trinkets/a42e5b43e8)
This example illustrates how to retrieve molecular properties of a list of CIDs. In this example, the three pieces of information required for a PUG-REST request (that is, the input, operation, and output) are stored in respective variables (Lines 12-14). These variables are used to construct a PUG-REST request URL, which is stored in a variable called “url” (Line 17). In Line 20, this “url” variable is used as an argument in the function “urllib.request.urlopen()”, which makes the PUG-REST request.
Note that Example 2 uses additional variables to store different parts of the PUG-REST request URL, making it longer than Example 1. It is possible to reduce Example 2 into a three-line script without using these additional variables, by directly specifying the PUG-REST URL in the parentheses after “urllib.request.urlopen” as shown in Example 1.
Example 3: 2-D similarity search using CID queries (https://trinket.io/library/trinkets/490a1841a3)
In this example, 2-D similarity search is repeatedly performed using a list of CIDs as queries. The query compounds are defined in Line 17. Because 2-D similarity search can take only one query compound at a time, it should be repeated using a “for” loop as shown in Lines 20 through 28. In this “for” loop, each CID in “queries” (defined in Line 17) is assigned to the variable “mycid”, which is used to construct a PUG-REST request URL in Lines 24-25.
Note that the input CID is converted into a string [using the str() function at line 24] before it is concatenated with the other parts of the URL. This conversion is necessary because the input identifiers are provided as numbers at Line 17. If they are provided as strings (with each of them enclosed in quotes), they could be directly used in the URL.
Example 4: 2-D similarity search using isomeric SMILES queries (https://trinket.io/library/trinkets/3437cdefb0)
This example also performs 2-D similarity search using a list of SMILES strings as queries. An important difference between Examples 3 and 4 is the way in which the input identifiers are encoded in the PUG-REST request URL. Some special characters used as input identifiers are also reserved for the URL syntax, causing issues when directly encoded in an URL path. An example is the “/” (forward slash) character used in isomeric SMILES and InChI strings. To avoid conflicts with URL syntax, these line notations need to be included in the options part of the URL (after the “?” mark) (as in Lines 27-28).
Programmatic Access to Web-Based Chemical Information – Examples
- Log in to post comments
References
(3) Kim, S. Expert Opinion on Drug Discovery 2016, 11, 843.
(4) Kim, S.; Thiessen, P. A.; Bolton, E. E.; Bryant, S. H. Nucleic Acids Res. 2015, 43, W605.
Questions
-
Formulate a PUG-REST request URL to perform the following tasks.
- Get the full record (in XML) for CID 60823 that are derived from its 3-D structure.
- Get the small-size 2-D image of CID 2244.
- Get the XLogP and TPSA values of CID 1983, 2244, and 3672 in the CSV format.
- Get the list of CIDs whose name contains the string “crestor” in the TXT format.
- Get the list of CIDs for compounds that are similar to CID 3348 in terms of 2-D similarity (at the threshold = 95)
- Get the full record (in XML) of CIDs that have the substructure “C3=NC1=C(C=NC2=C1C=NC=C2)[N]3”
- Get a list of AIDs (in TXT) that target the protein encoded by the KCNH2 gene.
- Get a list of CIDs (in TXT) for compounds tested to be active in AID 376.
- Get the bioactivity data (in CSV format) for AID 376.
- Get the sources of the boiling point annotations in PubChem (Hint: see Module 5).
- Get the full record (in XML) for CID 60823 that are derived from its 3-D structure.
-
This question is designed to test whether you can access PubChem data from a spread sheet program.
- Make a Google Sheet like this template (http://bit.ly/2o0pzoT) and auto-populate it with the CID, molecular formula, heavy atom count, and XLogP values for alkanes and alcohols (with 1 through 10 carbon atoms). Provide the link to the completed google sheet so that your instructor can access it.
- Compare the XLogP values of alkanes with those of alcohols. Which one is greater, and explain why.
- Make a Google Sheet like this template (http://bit.ly/2o0pzoT) and auto-populate it with the CID, molecular formula, heavy atom count, and XLogP values for alkanes and alcohols (with 1 through 10 carbon atoms). Provide the link to the completed google sheet so that your instructor can access it.
-
Modify the example python script 4 (https://trinket.io/python/3437cdefb0) to retrieve (in a CSV format) the molecular formula, molecular weight, hydrogen bond donor count, hydrogen bond acceptor count, and XLogP values of compounds returned from a 2-D similarity search using the following three smiles strings:
- C1CCNCC1
- C1CCC(CC1)(CC(=O)O)CN
- C1=CC=C(C=C1)NC2=CC=CC=C2C(=O)O
[Please create an trinket.io account, save your work in the trinket.io platform, and provide your instructor with the link to it.]
Comments 7
PUG-REST
No, but E-Utilities can do a range search
PUG-REST does not have a way to perform a property range search (e.g., retrieving all molecules with the molecular weight between 100 and 200). This kind of search is supported by another programmatic access route, called Entrez-Utilities (E-Utils). (Actually, almost everything you do using the Entrez system can be automated using E-Utilities, but E-Utils are limited to text or numeric fielded data (e.g., chemical name search and search by numeric value like molecular weight) and cannot be used for structure searches (super/substructure search, similarity search, ...) that take a chemical structure as a query (in the form of SMILES/InChI or provided through the chemical structure sketcher).
So, depending on the type of your tasks, you need to use PUG-REST, E-Utils, or some other programmatics access routs, although this module does not cover all access routes. If you have a particular task but you don't know what to use, feel free to contact me.
Sort of going the other way
Only computed properties are indexed.
Experimental Data
Google Sheets
You can't provide multiple chemical names as input.
It is possible to provide multiple CIDs as an input (in the form of a comma-seperated list) like this:
https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/cid/887,702,1031,263/property/MolecularFormula,MolecularWeight,CanonicalSMILES,HeavyAtomCount,XLOGP/CSV
However, you can't do this with chemical name inputs, because many chemical names contain commas in them (e.g., 1,2-dichloroethan), and the server cannot tell whether a comma is used as a separator, or as a part of the name. When chemical names are used as an input, you need to process one name at a time.